In a previous post I provided a statistical test to compare chimpanzee and human genomes. As you can read there, the post generated a very interesting discussion among the readers, and it seemed to me that the general feeling at the end was that my statistical method for performing genome-wide comparisons might have some merit, after all.
One reader suggested applying an identical test in order to compare two human genomes. That sounded like a very good idea to me, so I downloaded another human genome dataset from NCBI and performed a test.
For the benefit of readers, I’ll briefly recapitulate the simple comparison algorithm used in my previous test. 10,000 different sequences, each composed of 30 consecutive DNA bases (possible values: A, T, G and C) were randomly selected from chromosome N of genome A. A search for a matching pattern was then performed on the corresponding chromosome N of genome B. A pattern match was deemed to occur only when all 30 base pairs coincided perfectly – in other words, the head-to-head comparison between these DNA sub-strings was not relaxed, as occurs in many other tests in evolutionary comparative genomics. The total number of pattern matches found for that particular chromosome was then recorded. All chromosomes were tested in a similar fashion. Readers can view the latest results in the table and chart below, and compare them with the earlier results for the chimpanzee vs. human comparison.
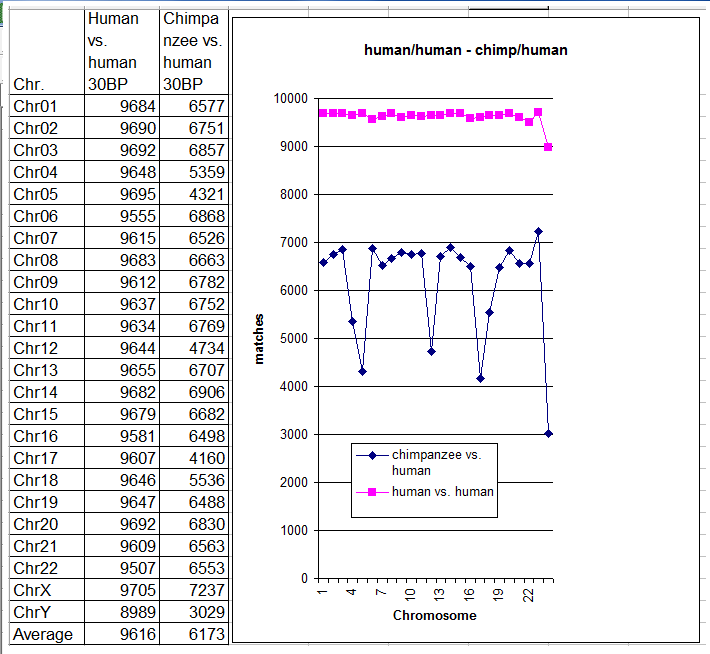
As expected, the number of pattern matches was always significantly greater when comparing two humans than when comparing a chimpanzee with a human. As the chart above clearly shows, the number of matches in human vs. human comparisons was quite stable, ranging from 9507 to 9705 (chromosome Y is the sole exception, with 8989). However, the same did not hold for chimpanzee vs. human comparisons, where the values were much more scattered.
Finally, the average number of pattern matches per chromosome, shown at the bottom of the table, was very different in the two cases: 9616 for human vs. human comparisons, but only 6173 for chimp vs. human comparisons. The average number of patterns without a match for human vs. human comparisons was (10000 – 9616) = 384, or in percentage terms, 384/10000 = 3.84%. The average number of patterns without a match in human vs. chimp comparisons was (10000 – 6173) = 3827, or in percentage terms, 3827/10000 = 38.27%, which is almost ten times greater.
So the bottom-line question is: if, as many evolutionists say, chimpanzee and human genomes are 99% identical, how “identical” are two human genomes?