
Typical figures published in the scientific literature for the percentage similarities between the genomes of human beings (Homo sapiens) and chimpanzees (Pan troglodytes) range from 95% to 99%. However, in press releases intended for popular consumption, evolutionary biologists frequently claim that human and chimpanzee genomes are 99% identical. Skeptics of neo-Darwinian evolution have repeatedly punctured this”99% myth,” but unfortunately, it seems to have gained widespread credence, due to its being continually propagated by evolutionists! For instance, one often encounters statements like these in the literature:
“Because the chimpanzee lies at such a short evolutionary distance with respect to human, nearly all of the bases are identical by descent and sequences can be readily aligned” (The Chimpanzee Sequencing and Analysis Consortium, Initial sequence of the chimpanzee genome and comparison with the human genome, Vol. 437/1 September 2005/doi:10.1038/nature04072).
“The consortium [National Human Genome Research Institute] found that the chimp and human genomes are very similar and encode very similar proteins. The DNA sequence that can be directly compared between the two genomes is almost 99 percent identical.” (here.)
“The genetic codes of chimps and humans are 99 percent identical.” (here)
Supporters of the neo-Darwinian theory of evolution have a strong ideological motivation for minimizing the differences between humans and chimps, as they claim that these two species evolved from a common ancestor, as a result of random mutations filtered by natural selection. Now, I don’t personally believe that humans and chimps share a common ancestry, for a host of reasons that would take me too long to explain in this post. Nor do I attach much significance to the magnitude of the genetic differences between these two species, per se, because in my opinion, the fundamental differences between these creatures lie elsewhere. However, since the genomic data is now available for free on the Internet, I decided to perform some sleuthing of my own, and check out the wildly exaggerated claims that are often made regarding the percentage similarities between human and chimp genomes. Here is what I discovered.
Interactive functional comparison methods
Usually, molecular biologists compare genomes on a functional basis. For example, they may search for similar genes in the genomes of human beings and chimpanzees, and try to identify the bases or nucleotides where they differ or match. Many different technologies have been developed to investigate genomes. One of these is BLAST (Basic Local Alignment Search Tool) software (see the NCBI Web site for more details). BLAST is an extremely powerful computer aided tool, as it is able to locate regions of local similarity among sequences by searching a whole database of genomes. Alignment methods (such as those implemented by BLAST and other techniques) allow geneticists to search interactively for common local patterns in different positions. However, this interactive task has its limits, as it can compare only portions of different genomes. Additionally, some critics have pointed out that these tools are susceptible to slip-ups (see here). Given the amount of data involved (in the order of Gigabytes), the global comparison of two genomes is a very demanding job, which cannot be completed interactively in a short time by human beings, even with the aid of tools such as BLAST. At the present time, only fully automated computer programs are capable of performing such a task on entire genomes. However, the development of an automated computer program which is capable of performing a complete functional comparison between human and chimpanzee genomes is practically impossible, for the simple reason that the functional architecture of these genomes is not yet perfectly known.
Automatic statistical comparison methods
From a mere informatics and statistical point of view, DNA sequences are simply strings of symbols or characters. Thus it is also possible to develop tests comparing genomes as unstructured sequences of characters, without taking into consideration genes, pseudo-genes, coding and non-coding regions, vertical and horizontal gene transfer, open reading frames (ORFs), or any other functional concepts. The characters most commonly present in DNA sequences are A, C, G and T. There are other less important characters which are used basically to indicate ambiguity regarding the identity of certain bases in the sequences. The comparison I performed was completely different from those usually performed by geneticists, because was purely statistical in nature. In a sense, it could be described as an application of the well-known Monte Carlo method. The Monte Carlo method is frequently used when data or processes involved are huge, and one wants to reduce the computer running time. In short, it involves dealing with a partial random sample, instead of the whole space which is under investigation. In the Monte Carlo method, only a small portion of the data population is actually investigated; nevertheless, this portion is statistically large enough to reveal the characteristics of the whole.
Metrics, distances and similarity measures
One theoretical approach to the problem would be to consider the set of all strings of characters as a metric space, and then define a distance function for all pairs of strings. Many distance functions have been developed by mathematicians for studying the degree of similarity between strings (for a list of them see (here). Given a metric or pseudo-metric space and its distance function, we can refer to a particular similarity, which differs from the similarity distance of another metric space. In a pairwise comparison identity test, we can easily calculate a simple metric distance called the “Hamming distance.” In this test, the order is important, because the n-th character of string A is compared to the n-th character of string B, after the initial characters of A and B have been aligned. After each comparison, if the two characters don’t match, then the Hamming distance increases by 1. If the order doesn’t matter, we can compare sub-strings of the parent strings A and B. Additionally, if they are at different positions in the two strings then many different tests are possible. We call these pattern matching or similarity tests. While there is only one possible method of comparing identity between strings of characters (the above pairwise comparison), there are many methods of comparing similarity. In other words, there are many measures of similarity, depending on the rules of pattern matching that we choose. In practice, calculating a certain distance function between two genomes can be a demanding job, in terms of running time, even for powerful computers.
Specifications for a statistical similarity test
Any final result for a complete statistical similarity test (especially if it is a unique number) is meaningful only if: 1) the distance function is mathematically defined; 2) the rules for pattern matching and the formulas for calculating the result are explained in detail; 3) it is clearly stated which parts of the input strings are being examined; 4) in the event that computer programs were used to perform the comparison, the source codes and algorithms are provided. My explanations below have the goal to meet the three first constraints. To satisfy the fourth condition, the source file of the Perl script used for the test is freely downloadable here.
How the genome data was obtained
Genome data for Homo sapiens and Pan troglodytes was freely downloaded from public bio-informatics archives at UCSC Genome Bioinformatics. The downloaded DNA sequences were in FASTA format. Before running the test, I decided to discard all symbols in the sequences, except for A, C, G and T. Most of the symbols I had to discard were “N” symbols, which represented rare, undefined situations (probably due to the level of sophistication of the scanning technology). The frequency of other symbols was very low. As it turned out, the deletion of a few “N” symbols didn’t affect the overall result very much. Given that the chimp’s genome contains two chromosomes (referred to as chr_02a and chr_02b) corresponding to chromosome #2 in human beings, I decided to concatenate them, in order to compare them with human chromosome #2 (chr_02).
30 Base Pattern Matching (30BPM) similarity test
The 30BPM similarity test is a very simple one: it performs searches for shared 30 base-long patterns, on two homologous chromosomes. This method is a true pattern-matching test, because it searches for identical patterns in the chromosomes of humans and chimpanzees. The beauty of this test is that it allows patterns to match, independently of their position in the chromosome. The significance of local similarities in homologous chromosomes is that identical patterns may be found in quite different positions along the two chromosomes. In fact, this test allows a total scrambling of patterns between homologous chromosomes. Of course, it is generally very difficult to know what the functional implications of this scrambling are. In particular, the positions of the genes might shift, but when non-gene coding is scrambled, it is doubtful that functionality is preserved. However, from a purely quantitative point of view, in this particular test, I don’t need to worry about qualitative issues such as functionality; only statistical issues count.
The algorithm implemented
For each pair of homologous chromosomes A and B, a PRNG (pseudo-random number generator) generates 10,000 uniformly distributed pseudo-random numbers which specify the offset, or starting point, of 10,000 30-base patterns that are contained in source chromosome A. The 30BPM test involves searching for all 10,000 of these DNA sub-strings of chromosome A in our target chromosome B. Now let F be the number of patterns located (at least once) in chromosome B. The 30BPM similarity is simply defined as F/100 (minimum value = 0%, maximum value = 100%). The absolute difference between 10,000 and F (minimum 0, maximum 10,000) is the 30BPM distance. Thus the greater the similarity is, the smaller the distance will be. Strictly speaking, this 30BPM space is only a pseudo-metric, inasmuch as the axiom of identity (“the distance is zero if and only if A and B are equal”) defining a true metric space is somewhat relaxed (in some cases, the distance could still be zero even if A and B were different), while the axiom of symmetry (“the distance between A and B is equal to the distance between B and A”) does not hold in some cases. It can easily be seen that the 30BPM distance will be zero (30BPM similarity = 100%) if the two strings are identical. In an additional test which I performed on two random 100 million-base DNA strings, the 30-BPM distance was 10,000 (i.e. no patterns on A were located in B). Hence I shall refer to the value 10,000 as the “random 30BPM distance.” In other words, the 30BPM similarity between two artificially generated random 100 million-base DNA strings is zero. Of course, when generating these artificial DNA strings I had to take into consideration the fact that that on average, the true probabilities of A, T, G and C occurring in natural DNA are not exactly 0.25 each, but as follows: A=0.3, T=0.3, G=0.2, and C=0.2. In such a case, the following formula accurately describes the probability of obtaining a single-base match between the two DNA sequences:
(30*30 + 30*30 + 20*20 + 20*20)/(100*100) = (900+900+400+400)/10000 = 26%
In a supplementary test in which I performed a pure pair-wise comparison between human/chimp genomes, I obtained a global figure 25.90%, which matches very closely with the theoretically predicted result above.
Results obtained
The following table and graph show the report of the 30BPM similarity test on the whole set of human/chimp chromosomes.
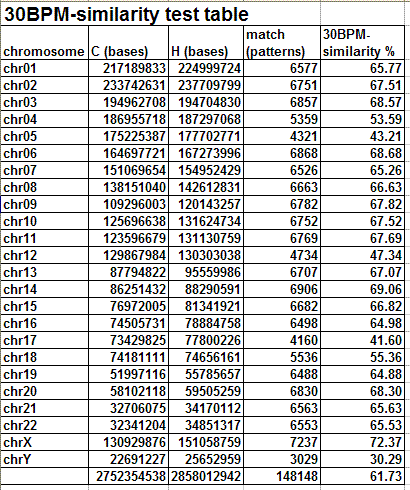
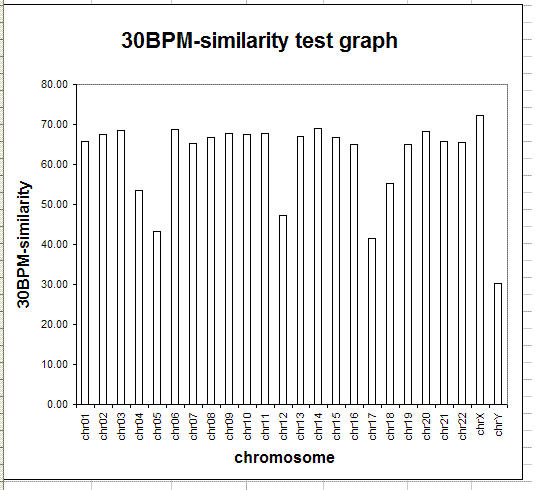
The results obtained are statistically valid. The same test was previously run on a sampling of 1,000 random 30-base patterns and the percentages obtained were almost identical with those obtained in the final test, with 10,000 random 30-base patterns. When human and chimp genomes are compared, the X chromosome is the one showing the highest degree of 30BPM similarity (72.37%), while the Y chromosome shows the lowest degree of 30BPM similarity (30.29%). On average the overall 30BPM similarity, when all chromosomes are taken into consideration, is approximately 62%. Here we have the classic case of the glass which some people perceive as being half-full, while others perceive it as being half-empty. When compared to two random strings which are 0% similar, 62% is a very large value, so nobody would deny that human and chimp genomes are quite similar! On the other end, 62% is a very low value when compared to the more than 95% similarity percentages which are published by bioinformatics evolutionary researchers. Now, I realize that it may seem somewhat arbitrary to choose 30-base-long patterns, as I did in my test, and indeed it is arbitrary to some degree. However, if the two genomes were really 95% similar or more, as is commonly claimed, also a 30BPM statistical test should produce 95% results, and it does not.
An analogy from politics: an exit poll
To help readers to grasp the significance and potential implications of my test, here is a simple analogy. Consider an election, in which 100 million electors are eligible to vote. One exit poll, based on a sample of 10,000 voters, calculates that party X has received 62% of the popular vote. However, at the end of election party X declares it has received more than 95% of the vote! The 30BPM statistical test described above is analogous to the exit poll, while the claims made by evolutionary biologists are analogous to party X’s “95%” claim. The sample of 10,000 patterns is taken from a global population of 100 million bases (the approximate number of bases on a typical human/chimp chromosome), while the ratio of population to sample is 100,000,000/10,000=10,000. The 30BPM exit poll metaphorically says that only 62% voted for Darwin’s party, whereas modern Darwinists claim that over 95% did. Something doesn’t quite add up.
I believe that the classic evolutionary comparisons between human and chimp genomes exaggerate the similarities, for at least two reasons: (1) they don’t consider whole chromosomes, but only portions of them (e.g. particular genes); (2) the rules of pattern matching are relaxed in some way (e.g. sometimes two bases are said to match, even when they don’t really match). Now, there is nothing intrinsically wrong with comparisons where (1) and (2) hold. However, any research that is truly worthy of being called “scientific” should openly acknowledge built-in limitations, such as (1) and (2) above. Sadly, this is very rarely done. It is perfectly acceptable to publish partial results that are obtained by relaxing the rules, but one should not publicize them as global and mathematically sound, when in fact, they are nothing of the sort.
Conclusion
We have seen that in a genome comparison, the only thing that matters is the degree of similarity. However, once we put the concept of similarity between two text strings on the table we open a can of worms. Many different measures of the similarity between two strings are possible, and different methods of comparing two genomes can result in wildly different estimates of the similarity between them. The assumptions that drive the methods used also drive the results obtained, as well as their interpretation. A simple layman’s statistical test, such as the 30BPM, shows that the “95% claim” described above is a highly controversial one. It is worth noting that as more information comparing the two genomes is published, the differences between them will appear more profound than they were originally thought to be. The big question that still remains is: what should one conclude from the similarities and differences between the genomes of humans and chimpanzees? Commonly reported evolutionary statistics that should provide an informative answer to this question may actually obscure the true answer.