 This morning I was observing some of the recent comment thread activity on Uncommon Descent, and my attention was drawn to this comment by Nick Matzke on the subject of the “onion test” argument for junk DNA:
This morning I was observing some of the recent comment thread activity on Uncommon Descent, and my attention was drawn to this comment by Nick Matzke on the subject of the “onion test” argument for junk DNA:
I have [The Myth of Junk DNA], and all [Jonathan] Wells does is gloss past T. Ryan Gregory’s onion argument; Wells gives the more important point, the huge variability in genome size as a widespread pattern, much attention at all. Considering Wells’s book is the definitive ID treatment of the junk DNA issue, and us ID critics have been bashing ID for its complete failure on the genome-size variability issue for years, this was a huge omission on Wells’s part.
Here, I offer a few thoughts on this fascinating subject.
What is the “Onion Test”?
Briefly stated, the “onion test” (which originates with T. Ryan Gregory) observes that onion cells have many times more DNA than we do. And since the onion is considered to be relatively simple as compared to the human, this discrepancy can only be accounted for within the context of the view that much of its DNA is, in fact, junk. This phenomenon is also known as the “C-value enigma”, and describes the lack of correlation (among eukaryotes) with respect to genome size and organismal complexity. The human genome comprises about 3 billion base pairs of DNA: Compare this to the genome size of Amoeba dubia (670,000,000,000 bp). Indeed, the human genome is only marginally bigger than that of C. elegans and D. melanogaster. In amphibians, the smallest genomes are just shy of 10 billion base pairs, while the largest are nearly 10^11 base pairs. Interestingly, the C-value paradox does not seem to apply to bacteria.
One Critical Assumption
This whole argument for junk DNA seems to rest on the critical assumption that having seemingly excessive amounts of repetitive DNA has no positive bearing on an organism’s physiology. But this assumption has been invalidated by the scientific evidence.
Transcriptional Delays and Timing Mechanisms During Development
One correlation which has been established is that highly-expressed genes tend to have short introns (Castillo-Davis et al., 2002), a likely reflection of the selective-pressure on transcriptional economy with respect to very highly expressed genes. Other genes are rich in introns: such as the 2400 kb human dystrophin gene, 99% of which is comprised of introns. The time taken to transcribe this gene into mRNA adds up to about 16 hours (Tennyson et al., 1995). To take another example, consider the Y chromosomal loci of Drosophila, which are extremely long — spanning millions of bases and consisting largely of introns. During the G2 phase of the primary spermatocyte (and only in that phase of that cell lineage) the Y chromosome unfolds to form species-specific nuclear architectures. A locus such as DhDhc7(Y) is transcribed over the course of two to three days to give rise to a ~5,100,000 nucleotide pre-mRNA (see Reugels et al., 2000; Piergentili et al., 2007; and Redhouse et al., 2011).
The time taken to transcribe respective stretches of DNA is not inconsequential to physiological fitness. Indeed, Swinburne and Silver (2010) explain,
Transcriptional delays were first invoked in 1970 while discussing biological timing for lambda phage and their use of long, late operons (Watson, 1970). Recognizing correlations between gene size and developmental timing, David Gubb later noted that the Drosophila Antennapedia (Antp) and Ultrabithorax (Ubx) genes owe their extreme lengths to large introns and formally introduced the intron delay hypothesis (Gubb, 1986). With the knowledge that the development of the fly’s body plan is sensitive to the proper expression of these genes in space and time, Gubb proposed that intron length could function as a time delay and aid the orchestration of gene expression patterns.
The paper further observes,
If intron delays have critical roles during developmental programs, then expression networks that depend on intron delays should be sensitive to perturbation of transcription elongation rates. Phenomena supporting this logic emerged in the genetic system of Danio rerio. The foggy and pandora mutants were identified for defects in both heart and neural development with the additional phenotype of shorter tails (Guo et al., 1999; Stainier et al., 1996). The mutants were mapped to the transcription elongation factors Spt5 and Spt6 (Cooper et al., 2005; Guo et al., 2000; Keegan et al., 2002). The nature of these mutants suggests critical roles for transcription elongation rates in the development of particular tissues and cell types. In the pandora (Spt6) background, researchers found that the transcripts of tbx20 (hrT), which encodes a protein required for heart development, are expressed inappropriately late during development and in the incorrect location when compared with wild-type (Griffin et al., 2000). While the molecular mechanism underlying this correlation might entail transcription initiation, elongation, RNA processing, or some combination thereof, the line of evidence suggests that transcriptional kinetics have important roles during vertebrate development.
Read the full paper for a list of further examples of this phenomenon.
Could Varying Genome Sizes Reflect Levels of Alternative Splicing?
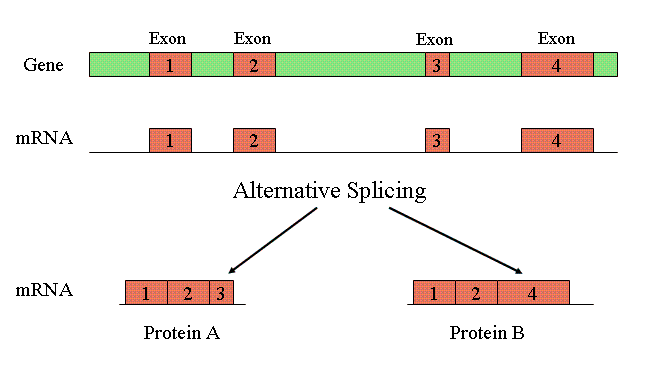
Perhaps some of the C-value enigma can be accounted for in terms of alternative splicing and alternative polyadenylation. Alternative splicing allows the exons of pre-mRNA transcript to be spliced into a number of different isoforms to produce multiple proteins from the same transcript, as shown in the diagram above. It is known that the level of alternative splicing exhibited in humans (about 90% — perhaps more — with an average of 2 or 3 transcripts per gene) is much higher than that for C. elegans (about 22%, with less than 2 transcripts per gene). This may, in part, explain why humans have only marginally more genes than C. elegans, which is otherwise seemingly paradoxical given the complexity of humans as compared to the roundworm. Moreover, bacteria do not undergo alternative splicing — which may, in some measure, explain their exemption from the C-value enigma.
Varying Preponderances of Transcription Factors
Approximately 10% of human genes code for transcription factors (a special class of protein which binds to specific sequences of DNA, namely, enhancers or promoters which are adjacent to genes which they regulate the expression of). In contrast, only about 5% of yeast genes code for transcription factors. When coupled with a much larger network of transcriptional enhancers and promoters, such a difference could result in a much larger set of gene expression patterns. This could lead to a non-linear increase in organismal complexity (see Levine and Tjian, 2003).
Are There Limiting Factors on Genome Size?
In 2002, Andrew George published a paper in Trends in Immunology, entitled, “Is the number of genes we possess limited by the presence of an adaptive immune system?” In the paper, he argued that the number of genes is limited in organisms which possess an adaptive immune system by the burden of self-recognition. As the paper explains,
The factors that are important in limiting the number of functional genes contained within the genome of an organism are presently unknown. Here, it is suggested that in organisms that contain an adaptive immune response, the number of genes in the genome might be limited by the need to delete autoreactive T cells, thus preventing autoimmunity. The more genes an organism has, the more autoantigens are generated, necessitating an increase in the proportion of T cells that are deleted.Is human complexity limited by the presence of an immune system? Although immunity is vital for health, the need to be tolerant to all ‘self’ molecules could restrict the number of genes in our genome.
A further correlation, which has been established, is that organisms with rapid development typically have lower C-values, presumably because they don’t have time to replicate lots of DNA between cell divisions.
In The Myth of Junk DNA, Jonathan Wells observes,
There is a strong positive correlation, however, between the amount of DNA and the volume of a cell and its nucleus — which affects the rate of cell growth and division. Furthermore, in mammals there is a negative correlation between genome size and teh rate of metabolism. Bats have very high metabolic rates and relatively small genomes. In birds, there is a negative correlation between C-value and resting metabolic rate. In salamanders, there is also a negative correlation between genome size and the rate of limb regeneration
In the case of bacteria, which have single replicons per chromosome, they face selective pressure to limit the accumulation of non-genic DNA which might make the replication times longer and thus slow rates of reproduction. This means that their genome size is correlated with gene number, and thus increases in proportion to structural and metabolic complexity.
Larger Cells Require More DNA
Take a look at the following graph, excerpted from Cavalier-Smith (2004):
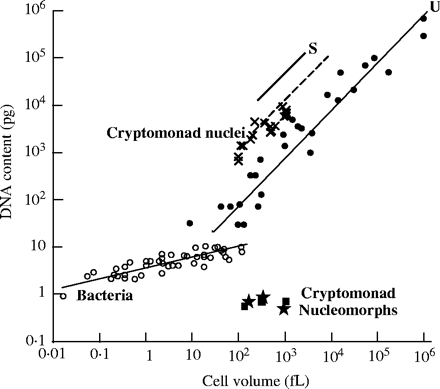
The graph shows a clear quantitative correlation between cell volume and DNA content. The trap into which the “junk DNA” advocate has fallen — as he so often does — lies with the (erroneous) assumption that all functions associated with DNA are sequence-dependent. But this need not universally be the case (in fact, it has long been shown not to be). This correlation holds not only true of vertebrate animals, but also for plants and unicellular eukaryotes (protozoa). It has been suggested by many that DNA possesses a structural role in controlling nuclear volume, cell size and cell-cycle length. Cavalier-Smith explains that, with increased cell size, “there is positive selection for a corresponding increase in nuclear volume; it is generally easier to achieve this by increasing the amount of DNA rather than by altering its folding parameters.”
As Thomas Cavalier-Smith observes,
Nuclear volume is probably functionally important for initiation of DNA replication and the transition from G1 to S: replication appears to initiate and terminate at the nuclear periphery and require a critical nuclear volume for onset (Nicolini et al., 1986); G1 nuclear volume growth must depend on concerted expansion of both chromatin and the nuclear envelope. But the significance of nuclear volume for the evolution of genome size does not depend on this, but on its fundamental significance for transcription, RNA processing and export, the rates of which must universally be adjusted to the rate of cytoplasmic protein synthesis. This unavoidable need for an optimal nuclear/cytoplasmic (karyoplasmic) ratio to allow balanced growth of actively growing and dividing eukaryotic cells means that larger cells must evolve proportionally larger nuclei. They can do that only by having larger genomes or unfolding DNA more; the former is mutationally much easier and quantitatively less limited and therefore predominates during evolution. Selection for economy means that smaller cells must have smaller nuclei. Mutations expanding or contracting the genome are always occurring with high frequency and will be selected long before any changing DNA folding patterns radically occur. Those are the fundamental reasons why genome size increases in larger cells and decreases in smaller ones. Bacteria, chloroplasts or mitochondria have no nuclear envelope attached to their DNA and no segregation of RNA and protein synthesis in two fundamentally different compartments; that is why their genome evolution follows different scaling laws: there is no selection for larger genomes in larger bacterial cells.
Summary & Conclusion
In summary, to point to the C-value paradox — or the so-called “onion test” — as evidence for the preponderance of junk or nonsensical DNA within animal genomes is based on several critical assumptions which are contradicted by recent data. The common naive supposition that having a larger genome size is neither here nor there in terms of organismal physiology has been shown to be untenable. With the ever-increasing expansion of our knowledge of the nature and functional inter-relatedness of the genome, those who choose to continue using the “junk DNA” argument as a club with which to beat intelligent design should find these facts disconcerting.