Jean Claude Perez is a self-organizational theorist, he is not a creationist. He has also published papers with an occasional visitor to UD, Andras Pellionisz.
If the mathematical/musical patterns Perez has found in DNA are improbable relative to laws of physics and chemistry, then he may have found yet another design feature of DNA, and this feature is found by combining coding DNA with non-coding DNA and viewing it holistically.
Here is the simplest explanation I found of his work:
When cells replicate, they count the total number of letters in the DNA strand of the daughter cell. If the letter counts don’t match certain exact ratios, the cell knows that an error has been made. So it abandons the operation and kills the new cell.
Failure of this checksum mechanism causes birth defects and cancer.
Jean-Claude Perez discovered an evolutionary mathematical matrix in DNA, based on the Golden Ratio 1.618
Dr. Jean-Claude Perez started counting letters in DNA. He discovered that these ratios are highly mathematical and based on “Phi”, the Golden Ratio 1.618. This is a very special number, sort of like Pi. Perez’ discovery was published in the scientific journal Interdisciplinary Sciences / Computational Life Sciences in September 2010.
Jean-Claude Perez discovered an evolutionary mathematical matrix in DNA, based on the Golden Ratio 1.618
Before I tell you about it, allow me to explain just a little bit about the genetic code.
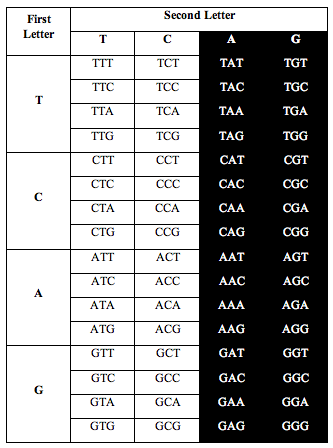
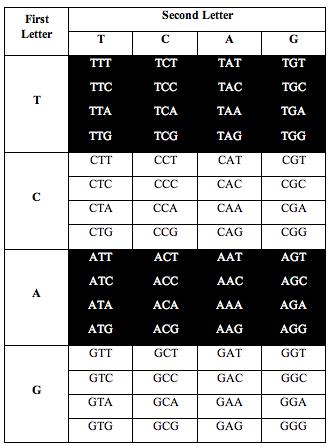
DNA has four symbols, T, C, A and G. These symbols are grouped into letters made from combinations of 3 symbols, called triplets. There are 4x4x4=64 possible combinations.
So the genetic alphabet has 64 letters. The 64 letters are used to write the instructions that make amino acids and proteins.
Perez somehow figured out that if he arranged the letters in DNA according to a T-C-A-G table, an interesting pattern appeared when he counted the letters.
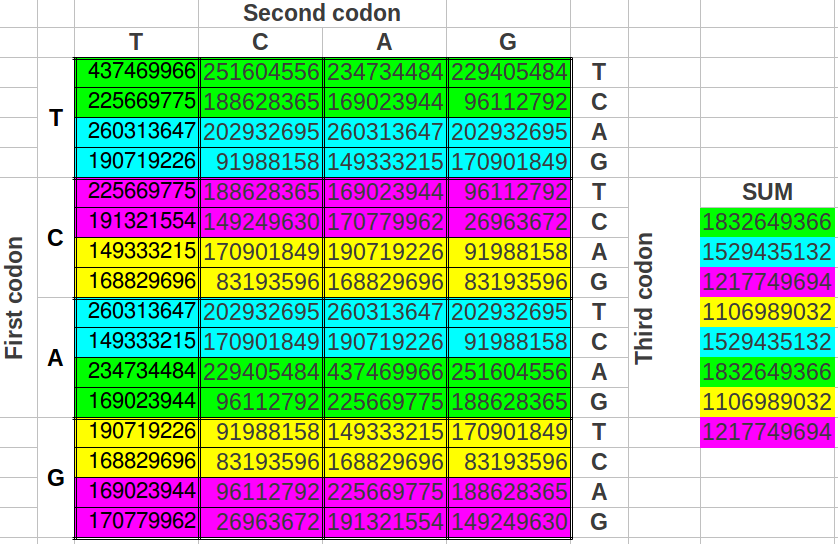
He divided the table in half as you see below. He took single stranded DNA of the human genome, which has 1 billion triplets. He counted the population of each triplet in the DNA and put the total in each slot:
When he added up the letters, the ratio of total white letters to black letters was 1:1. And this turned out to not just be roughly true. It was exactlytrue, to better than one part in one thousand, i.e. 1.000:1.000.
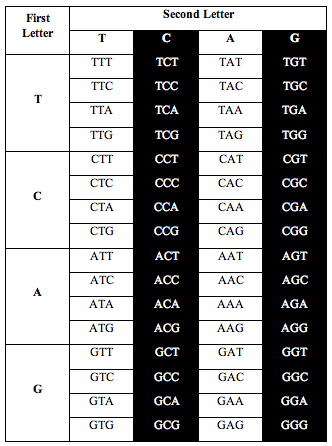
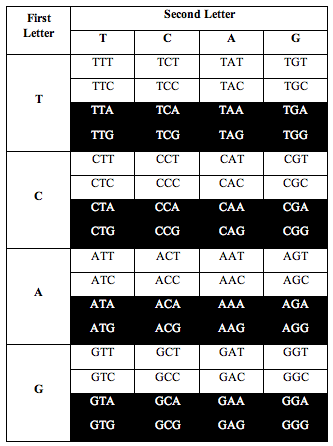
Then Perez divided the table this way:
Perez discovered that the ratio of white letters to black letters is exactly 0.690983, which is (3-Phi)/2. Phi is the number 1.618, the “Golden Ratio.”
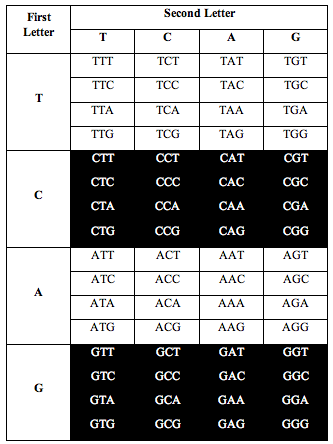
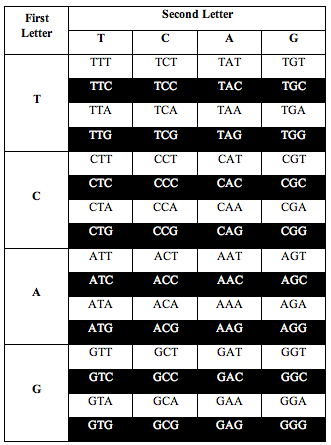
He also discovered the exact same ratio, 0.690983, when he divided the table the following two alternative ways:
Perez discovered that the ratio of white letters to black letters is exactly 0.690983, which is (3-Phi)/2. Phi is the number 1.618, the “Golden Ratio.”
He also discovered the exact same ratio, 0.690983, when he divided the table the following two alternative ways:
Above: Total ratio of white:black letters = 1:1
So for three ways of dividing the table, the ratio of white to black is 1.000:1.000.
And for the other three ways of dividing it, the ratio is 0.690983 or (3-Phi)/2.
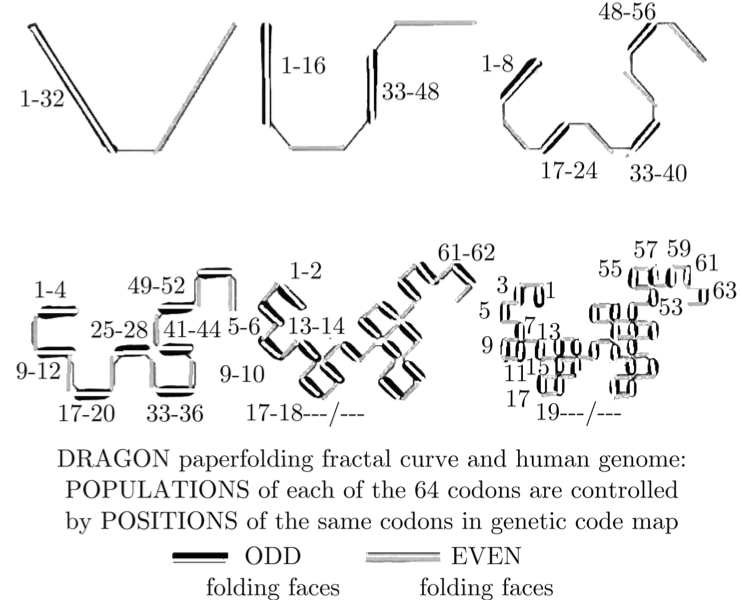
When you overlay these 6 symmetries on top of each other, you get a set of mathematical stairs with 32 golden steps. Then an absolutely fascinating geometrical pattern emerges: The “Dragon Curve” which is well known in fractal geometry. Here it is, labeled with DNA letters in descending frequency:
You can see other non-DNA, computer generated versions of this same curve here.
Other interesting facts:
•Similar patterns with variations on these same rules are seen across a range of 20 different species. From the AIDS virus to bacteria, primates and humans
•Each character in DNA occurs a precise number of times, and each has a twin. TTT and AAA are twins and appear the most often; they’re the DNA equivalent of the letter E.
•This pattern creates a stair step of 32 frequencies, a specific frequency for each pair.
•The number of triplets that begin with a T is precisely the same as the number of triplets that begin with A (to within 0.1%).
•The number of triplets that begin with a C is precisely the same as the number of triplets that begin with G.
•The genetic code table is fractal – the same pattern repeats itself at every level. The micro scale controls conversion of triplets to amino acids, and it’s in every biology book. The macro scale, newly discovered by Dr. Perez, checks the integrity of the entire organism.
•Perez is also discovering additional patterns within the pattern.I am only giving you the tip of the iceberg. There are other rules and layers of detail that I’m omitting for simplicity. Perez presses forward with his research; more papers are in the works, and if you’re able to read French, I recommend his book “Codex Biogenesis” and his French website. Here is an English translation.
(By the way, he found some of his most interesting data in what used to be called “Junk DNA.” It turns out to not be junk at all.)
OK, so what does all this mean?
•Copying errors cannot be the source of evolutionary progress, because if that were true, eventually all the letters would be equally probable.
•This proves that useful evolutionary mutations are not random. Instead, they are controlled by a precise Evolutionary Matrix to within 0.1%
•When organisms exchange DNA with each other through Horizontal Gene Transfer, the end result still obeys specific mathematical patterns
•DNA is able to re-create destroyed data by computing checksums in reverse – like calculating the missing contents of a page ripped out of a novel.No man-made language has this kind of precise mathematical structure. DNA is a tightly woven, highly efficient language that follows extremely specific rules. Its alphabet, grammar and overall structure are ordered by a beautiful set of mathematical functions.
More interesting factoids:
The most common pair of letters (TTT and AAA) appears exactly 1/13X as often as all the letters combined – consistently, the genomes of humans and chimpanzees.
If you put the 32 most common triplets in Group 1 and the 32 least common triplets in Group 2, the ratio of letters in Group1:Group2 is exactly 2:1. And since triplet counts occur in symmetrical pairs (TTT-AAA, TAT-ATA, etc), you can group them into four groups of 16.
When you put those four triplet populations on a graph, you get the peace symbol:
dna_peace_symbol
Does this precise set of rules and symmetries appear random or accidental to you?
My friend, this is how it is possible for DNA to be a code that is self-repairing, self-correcting, self-re-writing and self-evolving. It reveals a level of engineering and sophistication that human engineers could only dream of. Most of all, it’s elegant.
Cancer has sometimes been described as “evolution run amok.” Dr. Perez has noted interesting distortions of this matrix in cancer cells. I strongly suspect that new breakthroughs in cancer research are hidden in this matrix.
I submit to you that the most productive research that can possibly be conducted in medicine and computer science is intensive study of the DNA Evolution Matrix. Like I said, this is just the tip of the iceberg.
There is so much more here to discover!
When we develop computer languages based on DNA language, they will be capable of extreme data compression, error correction, and yes, self-evolution. Imagine: Computer programs that add features and improve with time. All by themselves.
What would that be like?
Perry Marshall

Elizabeth Liddle with her love of music might actually appreciate the possibility that life and musical notes are possibly intertwined.
I can’t do justice to the topic by quoting snippets, so I will have to provide a link:
Phi and Music DNA Here is diagram from that article:
Recall the discussion of ID and tuning of musical instruments here.
Perez bio is in creation wiki:
Jean-Claude Perez, Ph.D., is a French interdisciplinary scientist born on June 26, 1947 in Bassens, Gironde near Bordeaux (France). An engineer and French scholar from Bordeaux university[1], Perez worked principally with IBM in both the areas of Biomathematics and Artificial Intelligence (the first time, showing evidence of high level self-organization in cellular automata networks [2] and the second time creating neural networks with “FRACTAL CHAOS”(Fractal geometry), his holographic-like memory system and novelty detector). Then, in 1990, Jean-claude Perez published strong links between the world of fractals and numbers of the Fibonacci sequence which are based on the Golden ratio[3]. In this last area, with the “DNA supracode”[4], he proved that DNA coding for genes is structured by proportions related to Fibonacci numbers[5] [6] [7]. He verified this discovery in the field of the HIV genome by partnerships with Professor Luc Montagnier[8], the discoverer of the HIV virus. He has worked for 20 years in the fields of whole genome numerical analysis and numerical decoding of genes as coding or non-coding DNA sequences (as demonstrated particularly by the last publications: Five last publications/conferences ).
Particularly, in “Interdisciplinary Science” September 2010 issue, J.C. Perez published a peer-reviewed paper proving that the whole human genome codon populations are managed by a “DRAGON fractal paper folding curve” fine-tuned around the “Golden ratio”. Particularly, this main paper entitled “Codon populations in single-stranded whole human genome DNA are fractal and fine-tuned by the Golden Ratio 1.618.” shows that the Universal Genetic Code Table not only maps codons to amino acids, but serves as a global checksum matrix at the whole genome macro-structural scale.[9]Golden ratio.jpg
Draft of the paper Final published paper
A complete summary of J.C Perez’s research was published in Pellionisz A., Graham R., Pellionisz P., Perez J.: Genome Function of the Cerebellum: Geometric Unification of Neuroscience and Genomics. In: Manto M., Gruol D., Schmahmann J., Koibuchi N., Rossi F. (Ed.) Handbook of the Cerebellum and Cerebellar Disorders: SpringerReference (www.springerreference.com). Springer-Verlag Berlin Heidelberg, -1. DOI: 10.1007/SpringerReference_310386 2012-03-12 14:15:14 UTC. Details in [10] and full paper available in [11]In october 2013 jean-claude perez published a peer reviewed major article in [APPLIED MATHEMATICS]http://www.scirp.org/journal/am/ (BIOMATHEMATICS issue) entitled “The 3 genomic numbers discovery”. This article show – for the first way – that complete human genome single stranded DNA constitutes a WHOLE… [12]
http://creationwiki.org/Jean-claude_Perez
The “Vodka” designation means a highly speculative topic.