A few months ago, I posted a reasonably detailed introduction to the incredible molecular processes which undergird the activities of the bacterial flagellum — a remarkable high-tech rotory motor which confers motility to certain species of bacteria (the most studied being E. Coli and Salmonella). I posted this because the remarkable processes which I describe are not commonly discussed in these circles (which is somewhat ironic since we have made the flagellum our paradigm system). All-too-often those of a Darwinian persuasion are allowed to get away with the most outlandish of explanatory hypotheses in attempt to account for complex biochemical systems such as the flagellum. While these explanations may appear persuasive to the largely lay-audience, ill-aquainted with the sheer brilliance and design which undergirds these systems at the molecular level, closer inspection finds them wanting. Just as evolutionary “explanations” of the eye suddenly become inherently unpersuasive when one considers the remarkable biochemistry and molecular details of vision, so too do the purported “explanations” of the bacterial flagellum pale into triviality when one considers the biochemistry and molecular details undergirding its construction within the cell.
In this article, I want to take the opportunity to discuss in perhaps somewhat greater detail than I did previously, just how magnificent this system really is.
A Regulation Hierarchy

As I mentioned in my previous essay, the synthesis of the bacterial flagellum is orchestrated by genes which are organised into a tightly ordered cascade in which expression of one gene at a given level requires the prior transcription of another gene at a higher level.
In Salmonella, the flagellar system has three classes of promoters — Class I, Class II, and Class III. This sequential transcription is coupled to the process of flagellar assembly. Class I contains only two genes in one operon, namely FlhD and FlhC. Class II consists of 35 genes across eight operons. These genes include those involved in the biosynthesis of the hook-basal-body and other components of the flagellum and export apparatus, as well as the regulatory genes FliA and FlgM. Those genes which are involved in the synthesis of the filament are controlled by virtue of the Class III promoters.
The Class I promoter is required to drive the expression of the enteric master regulator, FldH4C2. The Class II promoters are subsequently turned on by this master regulator in association with the sigma factor, σ70. The Class II promoters are responsible for the gene expression of the hook-basal-body subunits and its regulators, including σ28 (encoded by a gene called FliA) and its anti-sigma factor, FlgM. The sigma factor σ28 is, in turn, required in order to activate the class III promoters. Before the construction of the Hook-Basal-Body has been completed, one obviously does not want the flagellin monomers to be prematurely expressed. Thus, in order to inhibit the σ28, its anti-sigma factor FlgM keeps it away from the RNA polymerase holoenzyme complex. When, finally, the Hook Basal Body has been completed, the anti-sigma factor FlgM is secreted, remarkably, through the flagellar substructures which are produced by the expression of the Class II hook-basal-body genes. The Class III promoters are then finally turned on by the sigma factor σ28, and the flagellum is completed. These Class III promoters are responsible for the expression of flagellin monomers, the chemotaxis system and the motorforce generators. In all, more than 50 genes are necessary for flagellar self-assembly to take place.
The Process Of Assembly
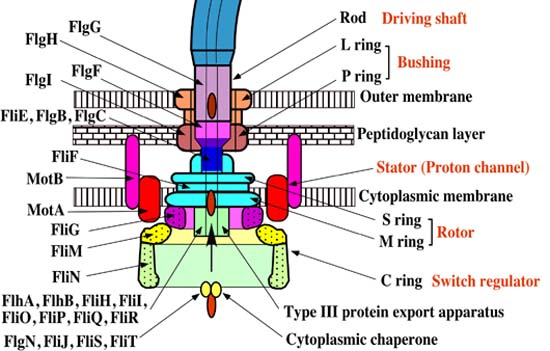
The flagellar apparatus can basically be divided into two key components: the secretion system and the axial structure. As discussed in my previous piece, the key components of the axial structure are FlgG for the rod, FlgE for the hook, and FliC for the filament. Each of those has its own respective cap protein, by virtue of which it assembles. The cap protein for FlgG is FlgJ; for FlgE, it is FlgD; and for FliC, it is FliD.
The cap protein FliD remains at the tip of the filament in the finished product. Some other components of the axial structure — FlgB, FlgC and FlgF — connect the rod and MS ring complex. The hook and filament are connected by FlgK and FlgL.
The structural foundation of the flagellar apparatus is the MS ring complex. When the C ring and C rod attach to the cytoplasmic surface of the M ring, the complex begins to secrete flagellar proteins.
One particularly remarkable feature of the flagellar assembly is the construction of the rod structure (which is built through the peptidoglycan layer with the assistance of cap protein FlgJ). The outer membrane represents a road block such that it cannot continue to grow. Incredibly, the outer ring complex actually cuts a hole in the membrane to allow the hook to continue to grow beneath the FlgD scaffold until it reaches a specified critical length, upon which the substrates which are being secreted switch from the rod-hook mode to flagellin mode. FlgD is then replaced by hook associated proteins (or HAPs) and the filament continues to grow. This, of course, only works correctly in the presence of the cap protein FliD; otherwise the flagellin monomers are lost.
Deeper And Deeper

What has been presented above is a somewhat shallow overview of the complexities of this magnificent process. So far, I have only skimmed the surface. We have observed the tip of the ice berg. The deeper one goes, however, the more it becomes clear that what one is looking at is not the product of the dual forces of chance and necessity, but the product of design. Let’s dig a little deeper.
As I alluded to above, with respect to Salmonella, regulation of transcription is contingent upon FlgM secretion at the time of the completion of the Hook-Basal-Body. FlgM itself is expressed from a master-regulator-dependent class II promoter and a σ28-dependent class III promoter. But here’s the thing. The translation of this FlgM from the class III transcript is itself subject to regulation. One protein (which is membrane-anchored) called Flk is responsible for positively regulating class III FlgM translation in L- or P-ring mutants. Another protein, called FlgN, is commissioned with the role of positively regulating class III FlgM translation, though this appears to be only in HBB mutants.
But it gets better. The flagellar export system (that is, the means by which FlgM is removed from the cell) has two substrate-specificity states: rod-/hook-type substrates and filament-type substrates. During the process of flagellar assembly, this substrate-specificity switch has to flick from the former of those states to the latter. Proteins which form part of the hook and rod need to be exported before those which form the filament. But how does this switch in substrate-specificity take place? The mechanistic basis of this switch possesses such a breath-taking elegance and ingenuity that a design argument could justifiably be built on this system alone – let alone the rest of the flagellum! Let’s take a peak.
Take a look at the following figure [source].
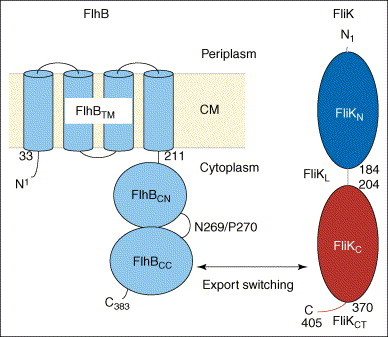
A membrane protein called FlhB, shown in the diagram in blue, is the key player in this process. Quoting one paper (Ferris et al., 2005),
One of the integral membrane proteins, FlhB, has been found to play a central role in export substrate-specificity switching; i.e. regulation of the order in which flagellar subunits are exported, such that proteins incorporated into the cell-proximal rod and hook structures (early export) are exported before proteins that polymerize to form the distal hook-filament junction and flagellar filament structures (late export) (16, 17). FlhB is a 42-kDa, 383-amino acid protein that has four putative transmembrane helices in its N-terminal domain (FlhBTM) and a sizable hydrophilic C-terminal domain (FlhBC)2 that is predicted to lie in the cytosol (17) (Fig. 1). The FlhBC domain itself is also divided into two subdomains: FlhBCN (amino acids 211–269) and FlhBCC (amino acids 270–383) connected via a proposed flexible hinge, based initially on the observation that overproduced soluble FlhBC is consistently and specifically cleaved within the hinge (N2692P270) with a half-life of ~5min, and the resulting subdomains remain tightly associated with each other so that they may be copurified (17). Site-specific mutagenesis of the highly conserved cleavage site sequence TN2PTH produced two variants, flhB(N269A) and flhB(P270A), each having a significant effect on FlhB function (16). The flhB(N269A) mutation completely inhibits cleavage, and the flhB(P270A) mutation slows it down significantly. Both mutant proteins are severely defective in their ability to mediate export of the “late” flagellar protein, FliC, whereas early flagellar protein export is unchanged. Cleavage of FlhBC is thus necessary for proper FlhB function. Previous studies also indicate that the cleaved state of FlhBC, rather than the cleavage event in itself, is necessary for substrate specificity switching (16). When FlhBTM+CN and FlhBCC are expressed from two different plasmids (resulting in an “already cleaved” FlhB) they are able to successfully complete both rod/hook and filament formation, although not to wild-type levels (17). All these observations led to a proposal that the C-terminal domain of FlhB has two substrate-specificity states and that a conformational change, mediated by cleavage and the interaction between FlhBCN and FlhBCC, regulates the specificity-switching process.

In addition, the flagellar hook-length control protein which is responsible for making sure that the hook length is of the right size (around 55nm) is called FliK. This same protein is also responsible for initiating the switch export substrate specificity. Several dozen of these molecules are randomly distributed in the cytoplasm. As it turns out, without FliK, both the ability to switch and export filament and the hook length control are completely lost.
One paper (Ferris and Minamino, 2006) describes the role of FliK thus:
The debate over the role of FliKN [the unstable N-terminal region from amino acid 1-184] in substrate-specificity switching encompasses two prevailing hypotheses that are currently being investigated – one proposes that FliKN is only necessary as an export signal to remove FliK from the cell (thus bringing it into contact with FlhBC) and thereby derepress hook substrate (FlgE) production; the other proposes that FliKN has the additional role of directly sensing hook-length during FliK export, and conveying this information by way of FliKC to FlhBC to result in the switch.
The paper goes on to present the following figure:
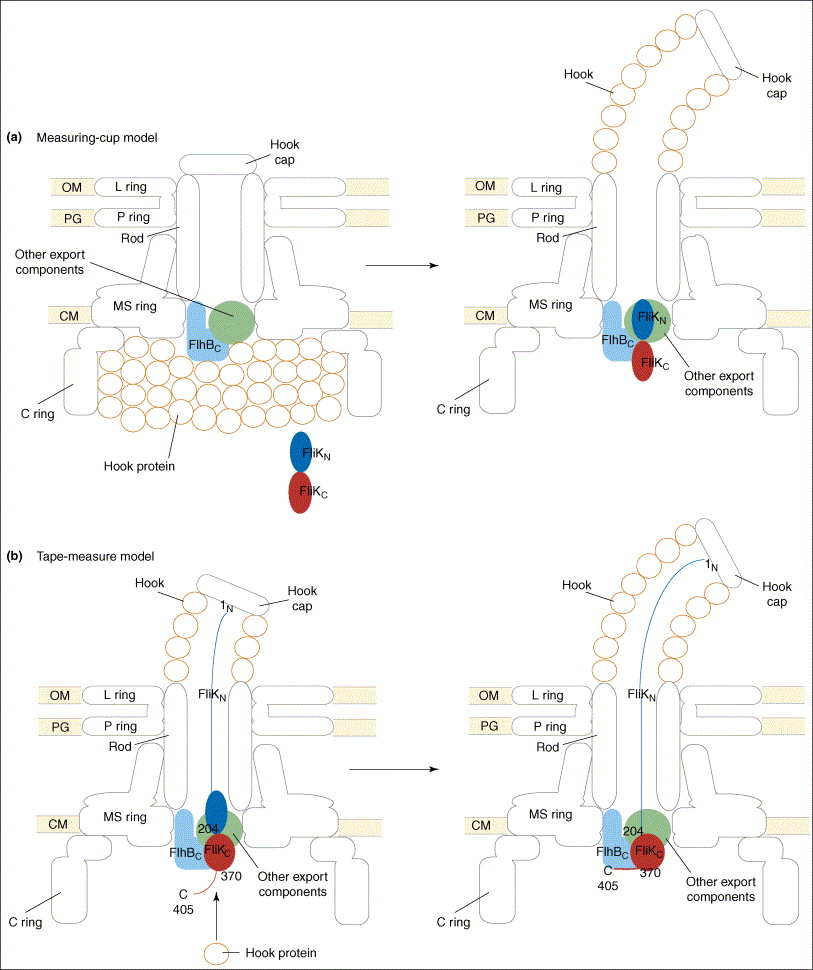
The figure legend explains,
Proposed models for the function of FliK in substrate-specificity switching. (a) The measuring-cup model. The cytoplasmic C ring of the flagellar basal body controls the length of the hook by functioning as a measuring cup that stores the hook proteins before their secretion. When the C ring is empty, FliK can interact with the cytoplasmic domain of FlhB (FlhBC), thereby changing the secretion specificity of the secretion apparatus. (b) The molecular tape-measure model. FliK consists of mainly two domains, the N- (FliKN) and C-terminal (FliKC) domains. During hook assembly, FliKN within the central channel of the hook-basal body functions as a molecular sensor and transmitter of information on the length of the hook. When the hook has reached its mature length, this information is transmitted to FliKC and FliKCT (370–405) to change their conformation to enable FliKCT (370–405) to bind to FlhBC, causing the conformational change in FlhBC that is responsible for the substrate specificity switch.
Conclusion

I’m still only scratching at the surface of this system. Time does not permit me to discuss all the features of flagellar assembly, and this post has not even covered the elegant molecular mechanisms underpinning chemotaxis, signal transduction, and rotational switching (discussed in my previous post in some detail). Indeed, cutting edge research in microbiology is still, in many respects, scratching the surface. There is still much we don’t yet know, and it is truly an exciting prospect for one to be involved in expanding our knowledge frontiers in this fascinating area.
As we learn more and more about these systems, the question inevitably arises, how did these complex and programmed systems come to be? Did they arise by design, or did they arise by unguided natural processes of chance and necessity? I leave you, the reader, to ponder this timeless question.
A wise and objective thinker once said,
If it could be demonstrated that any complex organ existed, which could not possibly have been formed by numerous, successive, slight modifications, my theory would absolutely break down.
Does the system by which the flagellum self-assembles not strike you as such a “complex organ”? Does it strike you as plausible that such a system could have been cobbled together one small step at a time while retaining a selectable element of utility all along the way? Perhaps such is the case, but the burden of proof must lie with he who subscribes to the affirmative, and not the other way about. Given the impotence of neo-Darwinism to account for novel protein folds and protein-protein binding sites, such a scenario seems totally unfeasible.
But there is a candidate explanation which seems to be staring us squarely in the face — design. Given that the selling point of Darwinism lies in its purported efficacy in explaining away design without recourse to a designer, shouldn’t design at least be considered as a possible alternative? Having read this piece (and, if you have time, my former one), watch Ken Miller present his “refutation” of Behe’s argument from irreducible complexity again and see if you are just as impressed as you were the first time.
Those hungry for further reading are directed to one of my favourite overviews of this topic: Here’s the Link.