Those of us who have been involved with the discussion and debate surrounding ID/evolution for any significant length of time will be quite aquainted with the most fashionable neo-Darwinian model for the origin of novel biological information: Gene duplication and divergence. Gene duplications normally arise from a phenomenon known as “unequal cross-over”, which occurs during cell division. This process results in the deletion of a sequence in one strand, and its replacement with a duplication from its homologous chromosome (meiosis) or its sister chromatid (mitosis). The model of gene duplication and divergence essentially maintains that, following a gene duplication, while one copy of the gene retains its original function, the other copy is freed from selective constraint and is thus free to mutate at a faster rate, and explore sequence space in search of some novel function.
A friend recently asked me to forward him some resources on this interesting and important subject, and thus I decided to write a brief article bringing together some of my thoughts on the subject.
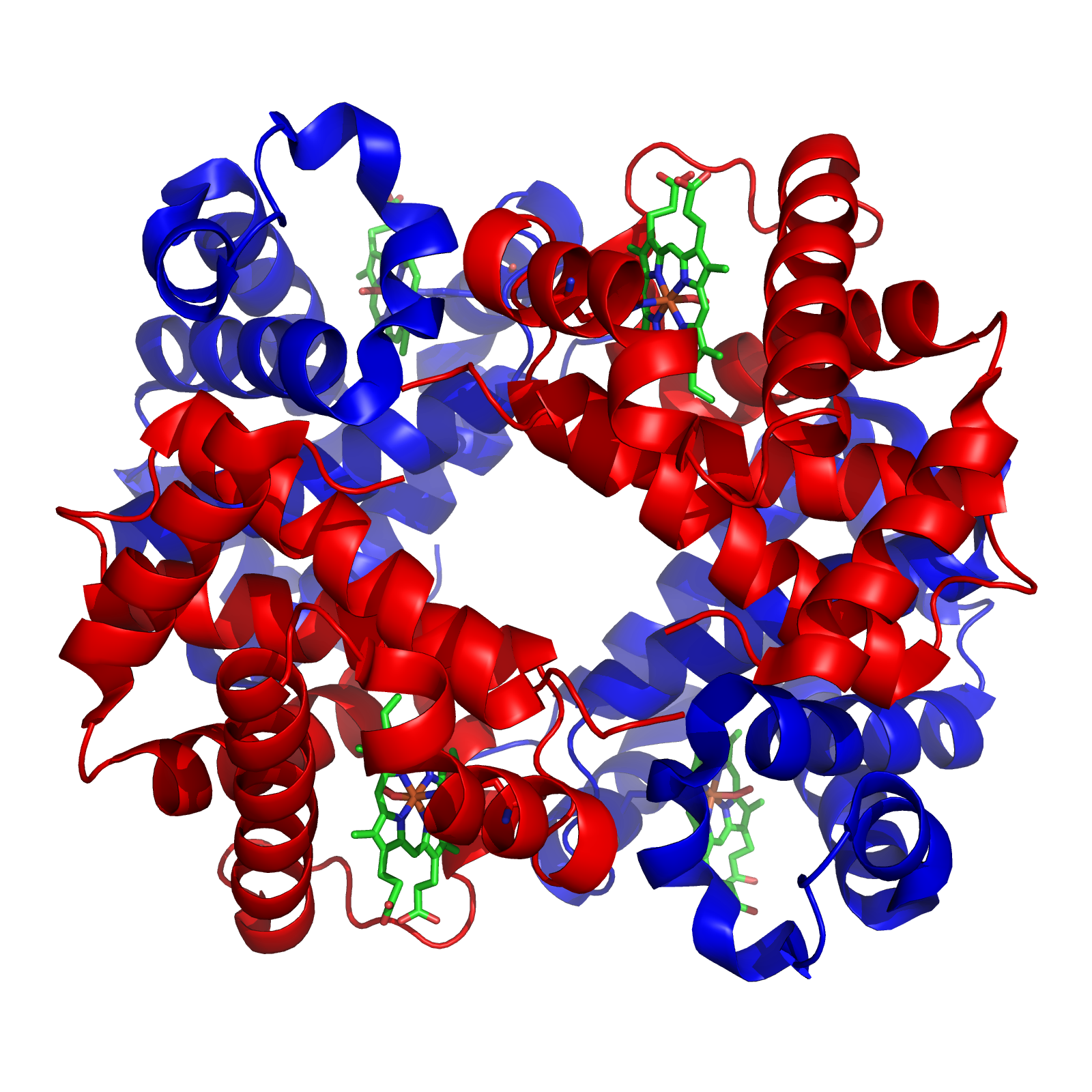 In this article, I wish to conduct a case study of the evolution of the globin family: haem-containing proteins which are characterised by their incorporation of the globin fold (a series of eight alpha helical segments). The globins are involved in the binding and/or transport of oxygen, the two best known examples being the reversible oxygen-binders haemoglobin (featured in the diagram to the left) and myoglobin.
In this article, I wish to conduct a case study of the evolution of the globin family: haem-containing proteins which are characterised by their incorporation of the globin fold (a series of eight alpha helical segments). The globins are involved in the binding and/or transport of oxygen, the two best known examples being the reversible oxygen-binders haemoglobin (featured in the diagram to the left) and myoglobin.
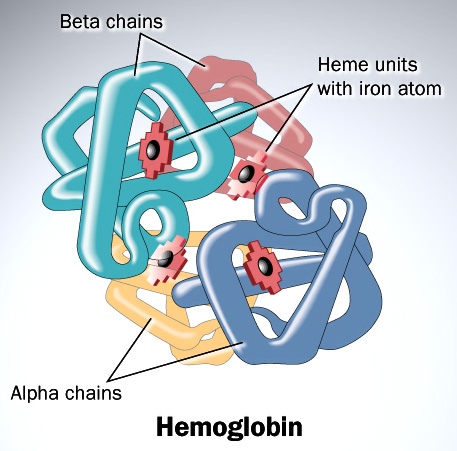
Haemoglobin is responsible for carrying oxygen from the lungs (or gills in the case of fish) to the tissues of the body where the oxygen is released to provide energy for the organism. Haemoglobin also serves to collect carbon dioxide to bring it back to the lungs so that it can be expelled from the organism’s body. Four polypeptide chains comprise the functional haemoglobin tetramer. Two of them are identical in structure (and are designated as the α-chain), and the other two are also identical with each other (and are referred to as a β-chain). In the diagram of the structure of haemoglobin (above), red designates the α subunits, whereas blue designates the β subunits. The iron-containing heme groups are coloured green. Myoglobin (which is specific to muscle cells) possesses an even higher affinity for oxygen than does haemoglobin. Myoglobin acts as a storage of oxygen, and retains it inside the heart and skeletal muscles.
Now, I submit that there are at least five difficulties associated with the evolution of the globins by virtue of gene duplication and divergence. These are:
- The question of the adaptive value of proposed intermediates.
- Complementary changes involving the regulation of gene expression.
- The time constraints associated with finding a selectable function for the duplicated copy.
- The fragility problem.
- Problems of convergence.
Let’s take a look at each in turn.
The Adaptive Value of Proposed Intermediates
 Consider the divergence between the α and β chains of haemoglobin. Following the duplication of the initial haemoglobin gene, each copy has to diverge simultaneously, and in complementary ways in order to ensure a functional tetramer. For one thing, characteristic of haemoglobin is that both chains possess hydrophobic residues which are essential for the association of the subunits (this stands in marked contrast to myoglobin which, being a water soluble protein, possesses mostly hydrophilic residues on the outside of its folded structure).
Consider the divergence between the α and β chains of haemoglobin. Following the duplication of the initial haemoglobin gene, each copy has to diverge simultaneously, and in complementary ways in order to ensure a functional tetramer. For one thing, characteristic of haemoglobin is that both chains possess hydrophobic residues which are essential for the association of the subunits (this stands in marked contrast to myoglobin which, being a water soluble protein, possesses mostly hydrophilic residues on the outside of its folded structure).
It is conventional wisdom that the early haemoglobin may have been a monomer (similar to myoglobin), which possessed exterior hydrophilic residues, some of which were subsequently substituted for hydrophobic residues. For example, in their book, Hemoglobin: Structure, function and evolution (1983), Richard Dickerson and Irving Geis argue that some of these substitutions occurred prior to the gene duplication: That is to say, the early haemoglobin evolved into a tetramer (or possibly a dimer) prior to its diverging into the α and β chains. Such a suggestion is, however, suspect in light of the fact that one might expect to see these hydrophobic amino acids in similar positions if they arose prior to the divergence of the haemoglobins: But the hydrophobic amino acids on the exterior of the polypeptides are different and are located in different positions on the α and β chains. This promotes their complementarity: Hydrophobicity is only needed at the sites that associate with the other chains (not necessarily at the same location on the different chains).
The α chains (unlike the β chains) are unable to make contact with one another. When the β chains are not produced (as in β-thalassaemia, a condition which is often fatal), the unassociated α chains typically degrade. In the case of α-thalassaemia, however, where it is the α chains which are lacking, the β chains do associate. This presents a different difficulty, for they bind oxygen with an affinity similar to that of myoglobin. This means that the oxygen will not be released to the tissues — again, with fatal consequences.
Given the plethora of pathological disorders which may be attributed to a change in just one amino acid in these chains (thus disrupting chain cooperativity), it should be quite evident that many amino acids in both chains are essential for the correct functioning of tetrameric haemoglobin.
Thus, as I hope to have shown above for our current case study (i.e. the globins), for gene family trees to be considered credible, it is necessary to show that there is a sufficiently high likelihood of all of the ancestral sequences conferring some sort of selective advantage. But it has been shown time and again that getting from one set of conserved amino acids to another — as is necessary for the production by divergence of proteins with different functions — is too big a jump through sequence space.
As documented by Axe and Gauger (2011), even a seemingly trivial switch from Kbl to BioF function requires at least seven co-ordinated mutations, putting the transition well beyond the reach of a Darwinian process within the time allowed by the age of the earth. Their paper studies the PLP-dependent transferases superfamily. They identified a pair within the superfamily with close structural similarity but no overlapping function. The enzymes chosen were Kbl (which is involved in threonine metabolism) and BioF (which is part of the biotin synthesis pathway). And they used a three-stage process to identify which sequences were most likely to confer a change in function.
This three stage process involved:
- Using structural and sequence comparisons of the two enzymes to identify candidate amino acids most likely to be functionally significant.
- Mutating these amino acids in BioF, making them like Kbl, and checking for loss of BioF activity.
- Testing whether changing these groups in Kbl to look like BioF would enable the Kbl to substitute for the function of BioF.
And thus they estimated that seven or more mutations would be required to convert Kbl to BioF function.
Axe and Gauger’s paper is not an isolated result. One review in Nature reported that changing an enzyme’s chemistry may require multiple neutral or deleterious mutations.
Interchanging reactions catalyzed by members of mechanistically diverse superfamilies might be envisioned as ‘easy’ exercises in (re)design: if Nature did it, why can’t we?…Anecdotally, many attempts at interchanging activities in mechanistically diverse superfamilies have since been attempted, but few successes have been realized.
When these results are taken into account in the context of the predictions of population genetics with regards the waiting time for multiple co-ordinated non-adaptive mutations which are required to facilitate a given transition (e.g. see Axe 2010), the situation for neo-Darwinism appears to be bleak.
Many readers will be aware of Douglas Axe’s famous 2004 JMB paper on β-Lactamase. Axe’s research set out with the initiative to ascertain the prevalence of sequence variants with a particular hydropathic signature which could form a functional structure out of the space of combinatorial possibilities. Axe began with an extremely weak (temperature sensitive) variant, entailing that an evolving new fold would be expected to be poorly functional. In so doing, Axe saught to detect variants operating at the lowest level — the threshold, if you will — of detectability. Axe sought to provide an estimate of the rarity of functional folds in all sequence space, which he gives as 1 in 10^77. This estimate was extrapolated from the number of variants which were able to carry out the function, no matter how weakly, of the TEM-1 β-Lactamase enzyme.
Axe’s work is not an isolated result either. For example, one study, published in Nature in 2001 by Keefe & Szostak, documented that more than a million million random sequences were required in order to stumble upon a functioning modestly sized ATP-binding protein. In addition, a similar result was obtained by Taylor et al. in their 2001 PNAS paper. This paper examined the AroQ-type chorismate mutase, and arrived at a similarly low prevalence (giving a value of 1 in 10^24 for the 93 amino acid enzyme, but, when adjusted to reflect a residue of the same length as the 150-amino-acid section analysed from β-lactamase, yields a result of 1 in 10^53). Yet another paper by Sauer and Reidhaar-Olson (1990) reported on “the high level of degeneracy in the information that specifies a particular protein fold,” which it gives as 1 in 10^63. I also strongly encourage readers to take a look at Douglas Axe’s excellent review article in Bio-complexity which covers this topic in more detail, as well as to read the recently-published The Nature of Nature — Examining The Role of Naturalism in Science, which is highly accessible for non-specialists. Axe has also published a blog post on the website of the Biologic Institute, addressing some criticisms of his 2004 JMB paper.
In Douglas Axe’s 2010 paper, he argues (on the basis of a bacterial population genetics model) that the gene duplication and recruitment, as a model for the evolution of new genes, is very limited. It works only if very few changes are required to reach a new selectable function. If the duplicated gene has a slightly negative fitness cost, the maximum number of mutations (in addition to the duplication itself) that a new innovation in a bacterial population can require is two or fewer. If the duplication is cost-free the number of mutations jumps to six or fewer.
Complementary Changes Involving the Regulation of Gene Expression
It often seems that evolutionary scientists are so interested in the similarity of structure and function of haemoglobin and myoglobin that they completely neglect the fact that they are produced in totally different tissues: bone marrow and muscle respectively. Myoglobin’s function is made possible by its higher affinity for oxygen. The modifications of the protein’s amino acid sequence, in order for it to be converted from haemoglobin to myoglobin, would have needed to be accompanied by complementary changes in its regulatory sequences in order to ensure that the myoglobin was produced in the muscle where it is needed, rather than in bone marrow where the red blood cells are produced. Myoglobin present in red blood cells would not provide a selective advantage. In fact, it would be harmful to the organism because it would bind too tightly to oxygen and not release it to the tissues (as in the case of α-thalassaemia described above). This is one of the most neglected points in discussions concerning gene duplication and family trees. Another example of this is the purported duplication of the β chain to give rise to the γ foetal version which possesses a higher affinity for oxygen. Such a scenario would, of course, have had to happen in concert with complementary changes in gene control such that it is expressed during pregnancy but synthesis of the β chain is suppressed until birth.
Time Constraints
It is widely believed that a duplicate gene has no phenotypic cost or advantage associated with it – that is, it is selectively neutral. In such a state, it is thought that the gene is free to mutate, independent of selection constraints or pressure. When a previously protein-coding gene incurs deleterious mutations such that it no longer codes for a useful polypeptide, the gene is rendered a “pseudogene”.
Kuo and Ochman (2010), however, in a paper published in PLoS Genetics entitled “The Extinction Dynamics of Bacterial Pseudogenes”, offer a potent challenge to this view. The researchers examine the genomes of Salmonella species, arguing that the evolution of bacterial pseudogenes is not an entirely neutral process. To the contrary, bacterial pseudogenes are often actively degraded by the forces of selection. Degradation of genes which do not code for proteins, or perform regulatory functions, is most probably due to the energetic costs of transcribing them into mRNA.
The researchers conclude that “Because all bacterial groups, as well as those Archaea examined, display a mutational pattern that is biased towards deletions and their haploid genomes would be more susceptible to dominant-negative effects that pseudogenes might impart, it is likely that the process of adaptive removal of pseudogenes is pervasive among prokaryotes.” They also suggest that the principle of pseudogene reductive (cost-cutting) evolution, might extend beyond the domain of prokaryotes, based on evidence for selection on the size of introns in some eukaryotic genomes (presumably, likewise, due to the energetic costs of transcription).
Many readers may recall the publication of a paper published last year in the journal, bio-complexity by Gauger et al. In that paper, a very similar conclusion was drawn. Gauger et al demonstrated that the process of reductive evolution was sufficient to prevent lines of E. coli from taking a simple two-step pathway to a new fitness function. In this study, a trpA gene was “broken” in such a way that it could recover the ability to synthesize the amino acid, Tryptophan, by reverting only two single point mutations. The authors here also concluded that the cost of transcribing a broken gene incurs significant fitness cost, and thus its removal is facilitated by positive selection.
What, then, can we conclude? Obviously, it supports the concept — which has been propounded by proponents of ID for some time — that there is a significant limitation on the number of mutations a “random walk” can accumulate prior to the degradation and loss of a ‘broken’ gene, thus significantly curtailing the ability of the mutational “search engine” to explore and survey the vast sea of combinatorial possibilities. It also serves as a potent reminder of the causal insufficiency of the Darwinian mechanism of random mutation and natural selection to account for the origins of fundamentally new protein structural domains and functions.
Indeed, as Kimura put it, in chapter 10 of his famous book, The Neutral Theory of Molecular Evolution (1983),
The process that facilitates the production of new genes will, at the same time, cause degeneration of one of the duplicated copies. In fact, the probability of gene duplication leading to degeneration must be very much higher than that leading to production of a new gene having some useful function.
The Fragility Problem
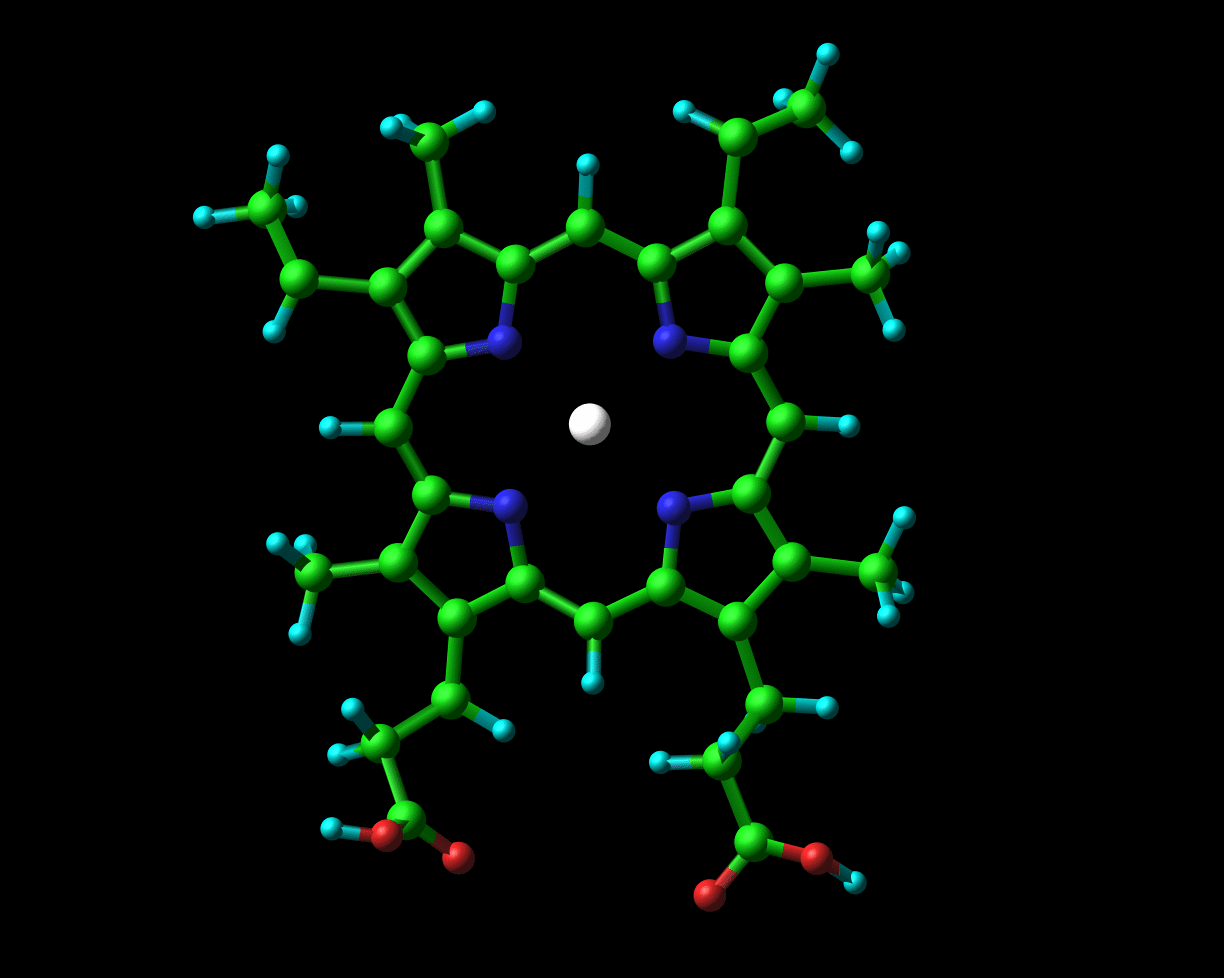 Globins possess a heme group (picture on the left) which contains iron. Iron exists in two oxidised forms, namely Fe2+ and Fe3+(ferrous and ferric forms respectively). When oxygen is available, the iron is readily oxidised to ferric Fe3+. But, crucial to its physiological role, the iron which is present in haemoglobin and myoglobin exists in the ferrous oxidation state. In chapter 2 of their book to which I previously alluded (Hemoglobin: Structure, function and evolution), Dickerson and Geis observe that “The purpose of the heme and the polypeptide chain around it is to keep the ferrous iron from being oxidized (metmyoglobin, with a ferric iron, does not bind oxygen), and to provide a pocket into which the oxygen can fit.”
Globins possess a heme group (picture on the left) which contains iron. Iron exists in two oxidised forms, namely Fe2+ and Fe3+(ferrous and ferric forms respectively). When oxygen is available, the iron is readily oxidised to ferric Fe3+. But, crucial to its physiological role, the iron which is present in haemoglobin and myoglobin exists in the ferrous oxidation state. In chapter 2 of their book to which I previously alluded (Hemoglobin: Structure, function and evolution), Dickerson and Geis observe that “The purpose of the heme and the polypeptide chain around it is to keep the ferrous iron from being oxidized (metmyoglobin, with a ferric iron, does not bind oxygen), and to provide a pocket into which the oxygen can fit.”
The ferrous iron, in haemoglobin and myoglobin, is actively prevented from being oxidised to the ferric form by the the specific chemical groups of the surrounding amino acid residues. Dickerson and Geis describe the fragility of this system as follows:
The met [oxidised] forms were experimentally the easiest to obtain, and the deoxy states also could be crystallized and studied with careful eperimental techniques. The oxy forms proved more intractable: Unless extreme care is taken, O2 oxidises the heme iron from Fe2+ to Fe3+ rather than simply binding, thus yielding the unwanted met form of the molecule.
As one might expect, the amino acids which surround the heme group are evolutionarily highly conserved. Moreover, many pathological conditions arise as the result of changing just one amino acid, resulting in the consequential inability of the polypeptide to retain the heme group correctly, thus permitting the iron to oxidise. In many cases, changing just one amino acid alters the positioning of the amino acids next to the heme group, such that they are no longer able to protect it from oxidation. This means that the altered amino acids need not even be in particularly close proximity to the heme group!
Problems of Convergence
While it is one thing to look at the sheer difficulty of globin evolution, the situation becomes even more bleak when one considers that the duplication to provide myoglobins and haemoglobins must have occurred at least twice in the jawless fish and in jawed vertebrates. From early on, it was concluded (from amino acid sequence comparisons) that the earliest globins were monomeric and thus led to the predominantly monomeric haemoglobins of the various invertebrate lines (e.g. insects, molluscs, jawless fish) and that only after this did the globin gene give rise to the separate myoglobin and haemoglobin of the jawed vertebrates. But the presence of myoglobin has been documented in lamphrey, a jawless fish (Romero-Herrera, 1979)!
There is, in fact, a number of examples like this. Another example is the alleged split resulting from the duplication of the α and β chains to give rise to the embryonically-expressed ζ and ε chains. In birds, one can find similar embryonically-expressed haemoglobin chains called π and ρ. It is widely thought that the divergence dates extremely anciently, preceding the split of the lineages leading to mammals and birds.
While the ζ and ε chains, though similar to each other, are markedly different from the α chains, modern ε and ρ chains exhibit far higher levels of similarity to mammalian and bird β chains respectively. It may thus be concluded that the split to give rise to these embryonic versions has occurred independently on at least two occasions — in each of the mammalian and avian lines.
Conclusion
In summary, we have seen that the scope for evolution of novel genes and proteins by virtue of gene duplication and subsequent divergence or recruitment is very limited, even in facilitating relatively trivial functional innovations. Given the extremely diverse array of protein conformations found in living systems, the likelihood of the relatedness of genes — even within gene families — may be treated with suspicion and healthy skepticism. It is somewhat ironic that biologists are all too willing to accept a statistical argument against two or more proteins with similar sequences arising independently by chance, but are completely unwilling to consider statistical arguments against them arising by chance at all.